Overview
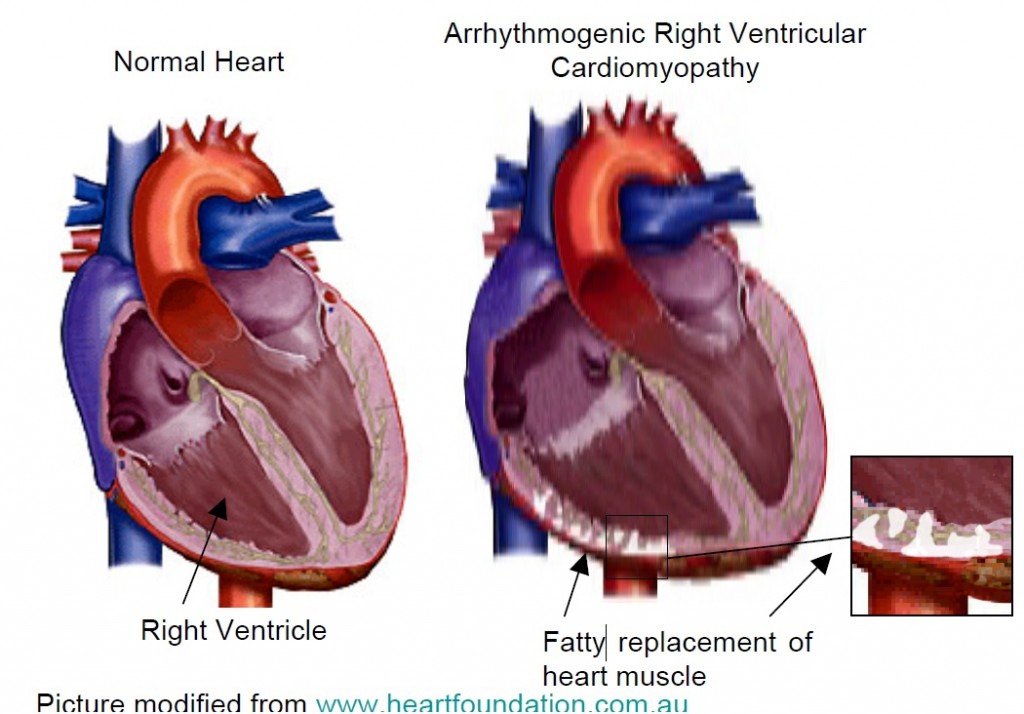
ARVC is a cardiomyopathy which affects both ventricles, but particularly the right ventricle. Detection in the early stages is difficult and in 1994 an International Task Force proposed criteria to aid diagnosis, which were revised in 2010. ARVC is characterised by a fibro fatty replacement of the heart cells of predominately the right ventricle (lower chamber of the heart), although changes can also be seen in the left ventricle. Genetic abnormalities called mutations have been found in the desmosomes, a protein that connect heart cells.
The condition is considered to be familial with autosomal dominant inheritance meaning, an affected individual has a 50% chance of passing the gene mutation on to their children but there are recessive types which are rare but can appear to skip generations. Arrhythmias (heart rhythm disturbances), especially ventricular tachycardia (fast heart rate from the lower chambers of the heart) are common and may cause sudden death. An implantable defibrillator is appropriate for sustained ventricular arrhythmias and may be considered in asymptomatic patients who meet task force criteria.
Management
Often a diagnosis of ARVC is difficult for doctors to make straight away and your doctor may ask for you to have several alternate investigations such as a special heart trace called a signal averaged ECG or a 24 hour heart monitor called a holter. When diagnosis is confirmed your doctor may advise medical therapy such as beta blockers, a medication to slow the heart rate and help to reduce arrhythmias. A preventative placement of an implantable defibrillator is recommended for some patients who meet the task force criteria. People with severe RV abnormalities may occasionally need heart transplantation. All first degree relatives of a person diagnosed with ARVC should be tested for this condition. ARVC may not be evident from the first set of tests you have, as it is a condition that can progress with age. Therefore we would advise that interval testing should be performed every few years.
Medical mode of diagnosis (Doctor Talk):
Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) occurs in approximately 1/5000 of the general population , but this is likely to be an underestimate. Diagnosis is made with a combination of two major, one major and two minor or four minor criteria. Studies of the right ventricle (RV) are a key investigation, best performed with an MRI although focussed echo may also be useful to help with diagnosis. Doctors will look for the presence of regional RV akinesia or dyskinesia with an end-diastolic volume ≥ 110ml/m² in males or ≥100ml/m² in females is a major criterion. Other major criteria include ARVC diagnosed in a first degree relative, symmetrical T wave inversion in leads V1-V3, LBBB morphology VT with a superior axis, or an epsilon wave which is a positive late deflection in lead V1 thought to represent delayed myocardial activation. Early accounts emphasized the “triangle of dysplasia” with involvement of the RV outflow, apex and inflow tract.
Inheritance and genetics:
Five desmosomal genes have been identified associated with ARVC, involving plakoglobin (JUP), desmoplakin (DSP), plakophilin-2 (PKP2), desmoglein-2 (DSG2), desmocollin-2 (DSC2) together with transforming growth factor beta-3 and TMEM43. Severely affected individuals may carry two mutations and caution is advisable in interpreting genetic testing as there are unknown mutations in some affected families.
The great majority of diagnosed individuals with appropriate management can expect a reasonably normal life span.